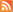
New Method Overcomes Drug Resistance in HIV/AIDS
5 Apr, 2007 04:54 pm




A major problem in HIV/AIDS is the virus ability to rapidly mutate which leads to the virus? escape from the body?s immune defense and the appearance of drug resistant strains. Even with the advent of new retardants and combination therapies the patient may ultimately develop resistance towards all available drugs. Since many new resistance strains constantly evolve in the population there is a great risk that current medications will become less effective over time.
Drug resistance in HIV/AIDS is due to that many mutant variations of the HIV virus are constantly formed. Some of these mutants may become insensitive to an HIV retardant. Hence the virus becomes resistant to the drug. The problem in HIV is that the virus produces very many variants which lead to many variations in the viral proteins. The Stanford HIV drug resistance database lists more than 24,000 HIV such variations for only one of the HIV proteins, called HIV protease. Very many of these variations cause resistance to the class of HIV retardants called protease inhibitors.
The role of the HIV virus protease is to cleave a protein chain, called polyprotein, into smaller fragments. The polyprotein is formed when the virus replicates, and after fragmentation of the polyprotein by the protease new HIV viruses are formed by the assembly of the fragments into mature infectious virons.
In a recent study we applied proteochemometrics to study the virus proteases ability to cleave many different protein fragments. Because proteochemometrics performs better when we include data from many different proteins we included not only HIV-1 proteases with many different mutations, but also proteases from eight other viral species, among them HIV-2, non-human immunodeficiency viruses, leukemia viruses, sarcoma viruses and others, all with many different mutations in them. In this way 61 mutated variants of proteases from 9 retroviral species was used to create a model for the ability of the proteases to cleave protein fragments; in total the model used up to 2162 such combinations of proteases and protein fragments.
We wanted to show that the model could be really used to predict the behavior of proteases. To show this we made a library in the computer of almost 2 ½ million protein fragments and used our model to predict how well they should be cleaved; the model then predicted that some fragments should be cleaved while others should not be cleaved at all. We selected 30 fragments; 15 were predicted to be non-cleavable, 7 were predicted to be cleavable with high rates, and 8 were predicted to be cleavable at a low rate. After that we manufactured the protein fragments in a special machine called peptide synthesizer, and tested how well they could actually be cleaved by different forms of drug sensitive and insensitive HIV-1 proteases.
The results from these tests were astoundingly good. The proteochemometric model predicted exactly which fragments were cleaved and which were not. Also the rates of cleavage were close to those that had already been predicted by the model in the computer.
We then used the proteochemometric model to analyze in detail the chemistry of viral proteases functions. This analysis revealed the chemical properties that are necessary for the proteases to cleave protein fragments and gave new insights into many of the inner chemical mechanisms working inside the proteases (see figure).
The proteochemometric technology is now in use to analyze the interactions of multiple drug resistant HIV-proteases with protease inhibitors with the aim to develop improved retardants that can inhibit a broad cluster of drug resistan HIV viruses. Studies are performed in collaboration with pharma industry as well as software dedicated for proteochemometric modeling is developed in collaboration with Genetta Soft, www.genettasoft.com.
References:
Kontijevskis A. et al. (2007) A look inside HIV resistance through retroviral protease interaction maps. PLoS Comput Biol 3(3): e48. doi:10.1371/journal.pcbi.0030048
Figure legend:
Functionally important amino acids in retroviral proteases revealed from the proteochemometric model. Shown in color on 3D structure models for the HIV-1 protease are amino acids of the protease that the proteochemometric model found to dynamically collaborate with amino acids of the protein fragment during its cleavage (the protein fragment is shown in the middle with its amino acids numbered P4 P4).
-
22/05/07
Preventing HIV Transmission to Infants Through Breastfeeding: The Key for Child Survival
-
04/05/07
New Drugs to Combat AIDS
-
03/05/07
DNA Damage Caused by Drug Used To Prevent HIV Transmission from Mother to Child
-
25/01/07
Cost-effectiveness of Adult Male Circumcision in AIDS prevention
-
11/01/07
HIV and Malaria: A Vicious Cycle

Interesting simulation.